ALL SMILES BEAT NF
Neurofibromatosis (commonly abbreviated NF; neurofibromatosis type 1 is also known as von Recklinghausen disease) is a genetically-inherited disorder in which the nerve tissue grows tumors (neurofibromas) that may be benign and may cause serious damage by compressing nerves and other tissues. The disorder affects all neural crest cells (Schwann cells, melanocytes and endoneurial fibroblasts). Cellular elements from these cell types proliferate excessively throughout the body, forming tumors; melanocytes also function abnormally in this disease, resulting in disordered skin pigmentation and café au lait spots. The tumors may cause bumps under the skin, colored spots, skeletal problems, pressure on spinal nerve roots, and other neurological problems.[1][2]
Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop. Therefore, if only one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well. The severity in affected individuals can vary, this may be due to variable expressivity. Approximately half of cases are due to de novo mutations and no other affected family members are seen. It affects males and females equally.
Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop. Therefore, if only one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well. The severity in affected individuals can vary, this may be due to variable expressivity. Approximately half of cases are due to de novo mutations and no other affected family members are seen. It affects males and females equally.
Neurofibromatosis type 1 (NF 1) Plexiform neurofibroma on the neck of a patient; plexiform neurofibromas are a cause of morbidity in the affected individuals. Main article: Neurofibromatosis type I Neurofibromatosis type 1 (also known as "von Recklinghausen disease"[1]) is the most common form of NF, accounting for up to 90% of all cases. NF 1 has a disorder frequency of 1 in 4,000, making it more common than neurofibromatosis type 2, with a frequency of 1 in 45,000 people.[3] It occurs following the mutation of neurofibromin 1 on chromosome 17q11.2; the large size of the NF1 gene (286kbp)[1] and presence of many homologous regions may predispose it to mutations. 100,000 Americans have neurofibromatosis. Neurofibromin is a tumor suppressor gene whose function is to inhibit the p21 ras oncoprotein.[3] In absence of this tumor suppressor's inhibitory control on the ras oncoprotein, cellular proliferation is erratic and uncontrolled, resulting in unbalanced cellular proliferation and tumor development. The diagnosis of NF1 is made if any two of the following nine criteria are met:
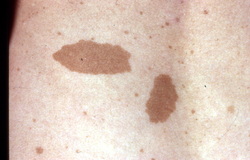
Patient with multiple small cutaneous neurofibromas and a 'café au lait spot' (bottom of photo, to the right of centre). A biopsy has been taken of one of the lesions.
Patient with multiple small cutaneous neurofibromas and a 'café au lait spot' (bottom of photo, to the right of centre). A biopsy has been taken of one of the lesions.
- Two or more neurofibromas on or under the skin, or one plexiform neurofibroma (a large cluster of tumors involving multiple nerves); neurofibromas are the subcutaneous bumps characteristic of the disease, and increase in number with age.
- Freckling of the groin or the axilla (arm pit).
- Café au lait spots (pigmented, light brown macules located on nerves, with smooth edged, "coast of California"[4] birthmarks). Six or more measuring 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals.
- Skeletal abnormalities, such as sphenoid dysplasia or thinning of the cortex of the long bones of the body (i.e. bones of the leg, potentially resulting in bowing of the legs)[1]
- Lisch nodules (hamartomas of iris), freckling in the iris
- Tumors on the optic nerve, also known as an optic glioma
- A first-degree relative (parent, sibling, or child) with NF-1 according to the preceding criteria.
- Scoliosis with or without kyphosis
- Macrocephaly in 30–50% of the pediatric population without any hydrocephalus[5]
- Epilepsy (seizures)
- Juvenile posterior lenticular opacity[1]
Neurofibromatosis type 2 (NF 2) Main article: Neurofibromatosis type II Neurofibromatosis type 2 (also called "central neurofibromatosis"[1]) is the result of mutation of the protein merlin (also known as "Neurofibromin 2" or "schwannomin"[1]) in chromosome 22q12. It accounts for only 10% of all cases of NF, and its frequency is lower than NF1. It is also caused by a mutation in a tumor suppressor gene NF2 (whose gene product is schwannomin or merlin). The normal function of merlin is not well understood.[3] The disorder manifests in the following fashion:
- bilateral acoustic neuromas (tumors of the vestibulocochlear nerve or cranial nerve 8 (CN VIII) also known as schwannoma), often leading to hearing loss. In fact, the hallmark of NF 2 is hearing loss due to acoustic neuromas around the age of twenty.
- The tumors may cause:
- headaches
- balance problems, and peripheral vertigo often due to schwannoma and involvement of the inner ear
- facial weakness/paralysis due to involvement or compression of the facial nerve (cranial nerve 7 or CN VII)
- patients with NF2 may also develop other brain tumors, as well as spinal tumors.
- deafness and tinnitus
